FamSeq
| Overview | |
| Description | FamSeq is a computational tool for calculating probability of variants in family-based sequencing data |
| Development Information | |
| GitHub | wwylab/FamSeq |
| Language | C++ |
| Current version | V1.0.2 |
| Platforms | Platform independent |
| License | GPL v3 |
| Status | Active |
| Last updated | 07/01/2014 |
| News | Version 1.0.2 includes option for GPU-based computing. |
| References | |
| Citation | Peng G., Fan Y., Palculict T.B., Shen P., Ruteshouser E.C., Chi A., Davis R.W., Huff V., Scharfe C., Wang W. Rare variant detection using family-based sequencing analysis. PNAS 110, p3985 (2013). https://doi.org/10.1073/pnas.1222158110 |
| Help and Support | |
| Contact | Wenyi Wang |
FamSeq
It is still challenging to call rare variants. In family-based sequencing studies, information from all family members should be utilized to more accurately identify new germline mutations. FamSeq serves this purpose by providing the probability of an individual carrying a variant given his/her entire family’s raw measurements. FamSeq accommodates de novo mutations and can perform variant calling at chrX.
FamSeq takes both likelihood and the widely used vcf files as input.
Download
Updated in v1.0.3: Allows for VCF output from FreeBayes.
Build and Run
1. Extract files from the compressed file
tar xvf FamSeq1.0.2.tar.gz
For windows user, please check here to extract the file.
2. Build FamSeq
CPU version:
cd FamSeq/src/
make
GPU version:
cd FamSeq/src/
make -f makefile.gpu
If you use MacOS 10.8 with XCode 5, or an error like “unsupported option ‘-dumpspecs’” occurs when compiling the GPU version, use the following command to compile
make -f makefile.gpu.clang
3. Run the test data
CPU version:
With VCF file as input:
./FamSeq vcf -vcfFile ../TestData/test.vcf -pedFile ../TestData/fam01.ped -output test.FamSeq.vcf -v
With likelihood only format file as input:
./FamSeq LK -lkFile ../TestData/loftest.txt -pedFile ../TestData/fam01.ped -output test.FamSeq.txt
GPU version:
With VCF file as input:
./FamSeqCuda vcf -vcfFile ../TestData/test.vcf -pedFile ../TestData/fam01.ped -output test.FamSeq.vcf -v
With likelihood only format file as input:
./FamSeqCuda LK -lkFile ../TestData/loftest.txt -pedFile ../TestData/fam01.ped -output test.FamSeq.txt
Documentation
Synopsis
FamSeq vcf -vcfFile input.vcf -pedFile input.ped -output output.vcf
FamSeq LK -lkFile lk.txt -pedFile input.ped -output output.txt
Commands and Options
First specify the command according to the input file type. If the input file is a VCF file, the command is vcf. If it is a likelihood only format file, the command is LK.
vcf
FamSeq vcf [-method 1] [-mRate 1e-7] [-v] [-a] [-l] [-vcfFile ] [-pedFile ] [-output] [-LRC] [-genoProbN] [-genoProbK] [-genoProbXN] [-genoProbXK] [numBurnIn] [numRep]
Options:
- -method integer
- The method used in variant calling. It is an integer. 1(default): Bayesian network. It works well when family size is less than seven. 2: Elston-Stewart algorithm. Use this method when family size is larger than 7 and the family has no loop. 3: MCMC.
- -mRate float
- Mutation rate. It is a float. The default value is 1e-7.
- -v
- Only record the position at which the genotype is not RR in the output file. (R: reference allele, A: alternative allele).
- -a
- Record all the positions in the output file. If there is an indel at one position, FamSeq will write the same line in input vcf file to output vcf file. The number of positions in input vcf file and output vcf file are the same. If option -v is set, option -a will be discarded. If neither ‘v’ or ‘a’ is set, FamSeq will record all the positions except the indel positions.
- -vcfFile string
- The name of input vcf file. All the individuals must be in one vcf file.
- -pedFile string
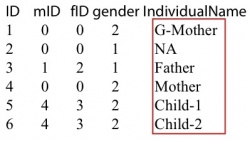
The name of ped file that store pedigree information. The pedigree should be a full family, which means that everyone in the family has two parents except for the founders of the family. There are five columns in the ped file.
Then we construct the corresponding ped file. Make sure the individual name in the ped file is the same as in the vcf file. The grandfather should be included in ped file with individual name NA, even though there is no information about him in the vcf/likelihood only format file. The file is shown on below: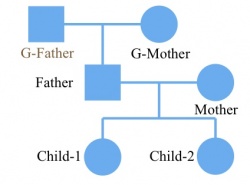 The first column is individual id that should be larger than 0. The second and third column is mother’s id and father’s id. If the individual is the founder of the family, set the mother and father’s id to 0. The forth column is gender. 1: male and 2: female. It will cause some errors at X chromosomes if the gender is not set correctly. The last column is individual name in the vcf/likelihood only format file. If there is no information of an individual in vcf/likelihood only format file, set the individual name to NA in the ped file. As the pedigree shown on the left. There are 6 individuals in this family. All individuals other than the grandfather were sequenced. Then the vcf file or the likelihood only format file look like the following:
The first column is individual id that should be larger than 0. The second and third column is mother’s id and father’s id. If the individual is the founder of the family, set the mother and father’s id to 0. The forth column is gender. 1: male and 2: female. It will cause some errors at X chromosomes if the gender is not set correctly. The last column is individual name in the vcf/likelihood only format file. If there is no information of an individual in vcf/likelihood only format file, set the individual name to NA in the ped file. As the pedigree shown on the left. There are 6 individuals in this family. All individuals other than the grandfather were sequenced. Then the vcf file or the likelihood only format file look like the following:
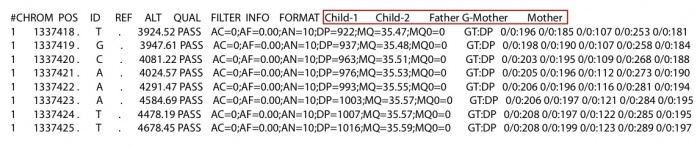

- -output string
- Output file name. If FamSeq calls a variant at a position, it will add two tags (FGT:genotype called by FamSeq and FPP: posterior probability estimated by FamSeq) at column FORMAT in vcf file.
- -LRC float
- A likelihood ratio cutoff. If likelihood (most likely genotype)/sum(likelihood of all genotypes) is less than the cutoff, we use pedigree information to improve variant calling. The default value is 1, we call all variant using pedigree information. Set it to 0 to only use single individual based method. Any values in between will determine whether FamSeq or single method is used for variant calling at a position.
- -genoProbN float float float
- Genotype probability of three kinds of genotype for autosome in population (Pr(G)) when this position is not in dbSNP. The default values are: 0.9985, 0.001 and 0.0005. The dbSNP position should be provided in column ‘ID’ in input vcf file.
- -genoProbK float float float
- Genotype probability of three kinds of genotype for autosome in population (Pr(G)) when the position is in dbSNP. The default values are: 0.45, 0.1 and 0.45.
- -genoProbXN float float
- Genotype probability of two kinds of genotype for chromosome X for male in population (Pr(G)) when the variant is not in dbSNP. The default values are: 0.999 and 0.001.
- -genoProbXK float float
- Genotype probability of two kinds of genotype for chromosome X for male in population (Pr(G)) when the variant is in dbSNP. The default values are: 0.5 and 0.5.
- -numBurnIn integer
- Number of burn in when the user chooses the MCMC method. The default value is 1,000n, where n is the number of individuals in the pedigree.
- -numRep integer
- Number of iteration times when the user chooses MCMC method. The default value is 20,000n.
LK
FamSeq LK [-method 1] [-mRate 1e-7] [-lkType n] [-v] [-a] [-l] [-lkFile ] [-pedFile ] [-output] [-LRC] [-genoProbN] [-genoProbK] [-genoProbXN] [-genoProbXK]
Options:
- -lkFile string
- Number of iteration times when the user chooses MCMC method. The default value is 20,000n.
- -lkType string
- The likelihood type. There are four types of likelihood: Normal (n), log10 scaled (log10), ln scaled (ln) and phred scaled (PS). The figure shown above is type n, without any scale.
- All other options are similar as in command vcf.
Output
FamSeq creates a new file by adding three columns to the original input file as the output file: GPP, FPP and FGT. GPP is the posterior probability calculated by single individual based method and FPP is the posterior probability calculated by FamSeq. These probabilities are all Phred-scaled. FGT is the genotype called by FamSeq.