Famdenovo
| Overview | |
| Description | Estimate the probability of deleterious germline mutations to be de novo using family history data. |
| Development Information | |
| GitHub | wwylab/Famdenovo |
| Language | R |
| Current version | 0.1.1 |
| Platforms | Platform independent |
| License | GPL v2 |
| Status | Active |
| News | Version 0.1.1 is now available |
| References | |
| Citation | A pedigree-based prediction model identifies carriers of deleterious de novo mutations in families with Li-Fraumeni syndrome. Gao F, et al. |
| Help and Support | |
| Contact | Wenyi Wang |
| Discussion | Issues On GitHub |
Famdenovo
Famdenovo is an algorithm that calculates the probability of de novo status in deleterious germline mutations using family history data. We demonstrate the utility of Famdenovo using deleterious TP53 mutations, which is a major cause for the Li-Fraumeni Syndrome , as de novo TP53 mutations account for 7-20% of the LFS patients.1,2
Supplementary Code for Gao et al. Genome Research 2020
R-markdown for how to use Famdenovo.BRCA scripts
R-markdown for figure making for the LFS cohorts
Download
Source File: Famdenovo_0.1.1.tar.gz . The source file is compatible with windows, linux and mac os.
For details about how to install and use Famdenovo, please check the Manual .
Installation
After downloading the R package, type the following command in the command line:
R CMD INSTALL Famdenovo_0.1.1.tar.gzOr type the following command in R Console to install the package:
install.packages("Famdenovo_0.1.1.tar.gz", repos = NULL)You can also install from GitHub:
library(devtools)
devtools::install_github("wwylab/Famdenovo/Famdenovo_0.1.1")Check here for details of R package installation.
Input Data Illustration
Famdenovo requires the following information as input: family, cancer, person.id, mutation, and gene.
Family Information Data
The input should be a data frame. The family data should include the following columns with the corresponding column names:
- id: index of the person. All individuals should be from one family.
- fid: index of the person’s father. If the individual is a founder of the pedigree, set it as NA.
- mid: index of the person’s mother. If the individual is a founder of the pedigree, set it as NA.
- gender: gender of the person. 0 - female; 1 - male
- age: age of the person. If the individual is alive, set it as the current age. Otherwise, set it as the age of death.
Example: For the pedigree shown below, pedigree information is stored as shown in the data file on its right:
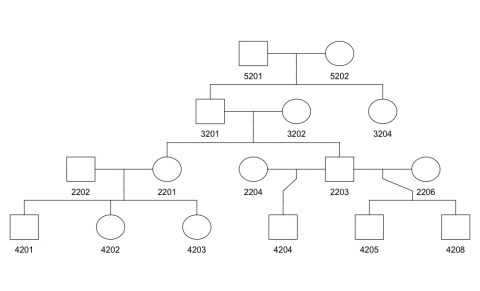
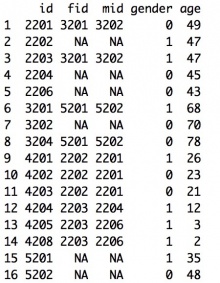
Cancer Information Data
The input should be a data frame. The cancer data should include the following columns with the corresponding column names:
- id: index of the person
- cancer.type: type of the cancer. We divided all cancers into 11 groups according to NCCCN Guidelines Version 1.2012 Li-Fraumeni Syndrome criteria. Check “LFSpro.cancer.type” for details.
- diag.age: The age when the individual was diagnosed with cancer.
Example:
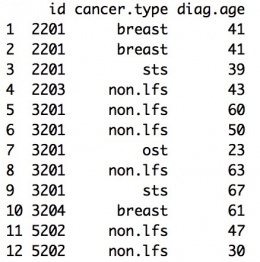
Mutation Information Data
The input should be a data frame. The mutation data should include the following columns with the corresponding column names:
- id: index of the person
- mut.state: mutation status of the person. “W” - wild type; “M” - mutated. Individuals who are not sequenced are not included in the mutation information data.
Example:
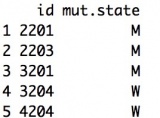
Person ID
The input should be either character string(s) or numerical value(s) of the person(s) you want to analyze.
Gene
The input should be character string(s). The default value is “TP53”. We have recently extended the Famdenovo function to BRCA1/2 as described above.
Package Illustration
Famdenovo(family, cancer, mutation, person.id, gene = "TP53")The following example briefly illustrates how to use the package.
library(Famdenovo)
# Test case 1
data(TP53.test1.family)
data(TP53.test1.cancer)
data(TP53.test1.mutation)
person.id <- c(2201, 2203, 3201, 3202, 4203)
output1 <- Famdenovo(TP53.test1.family, TP53.test1.cancer, person.id, TP53.test1.mutation)
output1
# Test case 2
data(TP53.test2.family)
data(TP53.test2.cancer)
person.id <- c(1, 11, 12, 13)
output2 <- Famdenovo(TP53.test2.family, TP53.test2.cancer, person.id)
output2Output
Currently we apply Famdenovo to the cancer gene TP53. The output of Famdenovo is the probability of a deleterious TP53 mutation being de novo.
# Test case 1
[1] "The following ids are not carriers: 3202, 4203"
id prob.denovo
1 2201 0.0001205471
2 2203 0.0001105599
3 3201 0.0126455556
# Test case 2
[1] "Warning: Famdenovo output is only applicatble to mutation carriers"
id prob.denovo
1 1 0.999994634
2 11 0.003633958
3 12 0.003774075
4 13 0.100231438Shiny Web App
The Shiny App for Famdenovo is also available. It is an interactive web app using the same inputs as the R software does. The output contains a pedigree structure of the family, the cancer information, and the de novo probability of mutation carriers calculated by Famdenovo.
- Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 46:689-93, 2009. http://dx.doi.org/10.1136/jmg.2008.058958 [return]
- Peng G, Bojadzieva J, Ballinger ML, Thomas DM, Strong LC, Wang W., Estimating TP53 Mutation Carrier Probability in Families with Li-Fraumeni Syndrome Using LFSPRO. Cancer Epidemiology, Biomarker and Prevention Jan 2017. https://doi.org/10.1158/1055-9965.EPI-16-0695 [return]