MetaBatch
| Overview | |
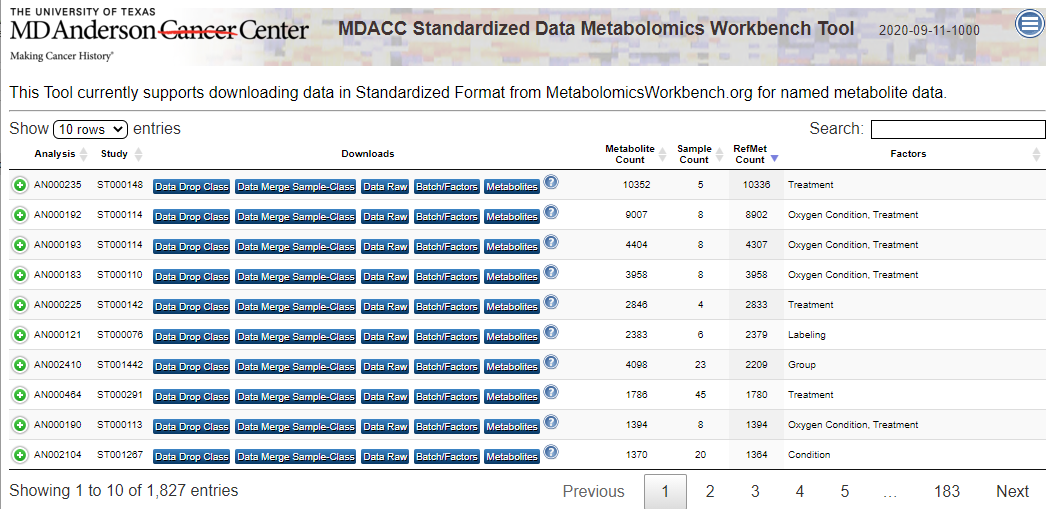
| Description | MetaBatch Web - Standardized Data for Metabolomics Workbench |
| Development Information | |
| GitHub | MD-Anderson-Bioinformatics/StandardizedData/ |
| URL |
https://bioinformatics.mdanderson.org/StdMW/ https://bioinformatics.mdanderson.org/MOB/ |
| Language | D3 (JS), Java |
| Current version | 2020-09-11-1000 |
| Platforms | web |
| Status | Active |
| Last updated | 2020-09-11 |
| News | Standardized Data access to Metabolomics Workbench Data |
| Help and Support | |
| Contact | Rehan Akbani |
MetaBatch
Description
The MetaBatch Standardized Data for Metabolomics Workbench app accepts input files from the world’s largest metabolomics data repository, Metabolomics Workbench (NIH) (MW). MW currently contains 464 MSI-compliant datasets representing numerous MS and NMR instrument platforms. The figure below shows the proposed architecture of the MetaBatch resource and how it will connect to public or private metabolomic databases.

Below is a link to sample Metabolomics Workbench Data processed by the in-development MetaBatch Web Analysis tool and as early proof-of-concept for the pipeline workflow described below.
VIEW EXAMPLE METABOLOMICS WORKBENCH DATA ANALYSIS
LAUNCH STANDARDIZED DATA FOR METABOLOMICS WORKBENCH WEBSITE
LAUNCH METABATCH OMIC BROWSER WEBSITE (ANALYSIS RESULTS)
MetaBatch architecture overview with three main components: (i) the database adapter that connects to public or private metabolomic databases; (ii) the computation API that performs assessment, detection and correction of batch effects; (iii) the visualization API for visualizing results using a repertoire of different dynamic plots.
See also the MBatch R package.
MetaBatch Workflow
The Standardized Data for Metabolomics Workbench Pipeline and MetaBatch Pipeline are two sides of the quality assurance coin. The Pipeline pulls publicly available deidentified clinical, batch-related, and -omic data from the Metabolomics Workbench and provides that data in a standardized format ready for analysis. The Batch Effects Pipeline uses Standardized Data to produce period runs of the MBatch R package with analysis and examples of correction runs. Both pipeline results are served via applications using Java, JavaScript, and HTML5 from web servers in Docker images.
The Pipeline consists of Java code running within Docker Linux containers that use the Metabolomics Workbench HTTP/RESTful API. The Pipeline checks for updated or new datasets and downloads a list of samples and files associated with the changes.
As in the approach we employed for TCGA, we will automate MetaBatch processing to enable the analysis of quantitative metabolomic data generated from multiple sources, including MS and NMR. We will also work with the curators of Metabolomics Workbench to develop a direct interface that allows MetaBatch to be run automatically after a data set is deposited in the Workbench repository. For other sites (e.g., core facilities), MetaBatch will be configurable for automatic or on-request operation as needed. The figure below shows a typical workflow diagram for the most common use cases.

MetaBatch Web
We will introduce MetaBatch in the form of a computational resource (tool) and also a web portal. The system will allow users with no programing experience to analyze their own data in an interactive graphical environment that enables the application of novel, quantitative measures of data quality and batch effects. Data entry and selection of analysis parameters will be achieved through an intuitive, aesthetically appealing interface. The visualization component will display dynamic, interactive plots with flexible user control. Corrected and uncorrected data will be available for download or for upload to the cloud. We will develop a user-friendly, “ready-to-run” version of the MetaBatch system that non-informatics users can easily install, configure, and use without IT services. It will be “run-anywhere” to avoid the need to support multiple operating systems and user environments. It will be based on Linux container technology using Docker. We were early bioinformatic adopters of Docker and have implemented it successfully for other projects (e.g., MBatch and NG-CHMs). For users who prefer to download and install MetaBatch on their own machines, we will create an install wizard to enable Macintosh and Windows users to run local instances of the MetaBatch Docker image without expert assistance. In accord with the RFA and Aim 5, we will obtain feedback on the usability of and workflow compatibility of MetaBatch by serial testing as well as by monitoring user forums.
MetaBach Algorithms
- Replicates-Based Normalization (RBN). RBN is a novel algorithm developed by PD/PI Rehan Akbani. It uses technical replicate samples from multiple batches to assess batch effects. It then subtracts those batch effects from the dataset by adjusting the location and scale of each metabolite on a per-batch basis. A major benefit of RBN is its capacity to merge multiple batches even when samples across the batches are very different and heterogeneous (e.g., from different tumor types).
- Empirical Bayes++ (EB++) (novel algorithm). EB++ (developed by PD/PI Akbani and collaborators) is a combination of empirical Bayes (ComBat [9]) with RBN that merges the strengths of the two. ComBat (i.e., EB) assumes that the batches have homogeneous samples and that any observed variation between batches is due to a batch effect, which it adjusts. Unfortunately, if two batches represent different tumor types, for example, ComBat will remove biological variation along with technical batch effects. RBN mitigates that problem by using technical replicates to assess batch effects directly. However, it can make only linear adjustments. ComBat can make non-linear adjustments and use priors for better adjustment of the data. EB++ uses replicates to estimate the batch effects and learn a “model,” which is then used to adjust the rest of the non-replicate data in a non-linear way, while taking advantage of prior information. Therefore, EB++ can mitigate the limitations of both ComBat and RBN. However, EB++ can represent over-kill when batch effects are linear.
- Distance Weighted Discriminant (DWD). DWD is a popular batch effect correction algorithm related to support vector machines, but with better performance for batch effect correction in high-dimensional data.
- Surrogate Variable Analysis (SVA). SVA is a useful batch effect correction algorithm that combines singular value decomposition (SVD) with linear model analysis [10].
MetaBatch Visualization
- Interactive, dynamic box plots. Box plots are extremely useful for visualization of data. We will provide interactive, dynamically explorable box plots that allow users to zoom, pan, and highlight the boxes for detailed investigation. Zooming in far enough will allow the distribution of points within the box to be viewed. Right-clicking (Windows) or control-clicking (Mac) on an individual point will open a submenu with interactive annotations (e.g., batch ID, number of data points represented). ANOVA p-values for statistical significance will be included. The box plots (and violin plots) will be scalable. Static Metabolomics Workbench Data Image: “/public-software/metabatch/Boxplot/BoxPlot_Group-Mean_Diagram-TIMEPOINT.png”
- Multivariate analysis of variance (MANOVA). We will implement MANOVA to test the association between different dependent batch variables (e.g., batch ID, tissue source site, and shipping date in the case of TCGA). To provide a graphical view of the impact of each batch variable, we will use our VennMaster implementation to provide Venn diagrams in which each oval represents the batch variable, the size of the oval represents the fraction of variance explained (FVE) by the variable, and the overlap represents the co-dependence.
- Principal Variance Component Analysis (PVCA). PVCA combines the strengths of PCA and variance components analysis (VCA). It provides a breakdown of the key sources of variation and quantifies them. Unexplained variation is called “residual.” Ideally, the variation associated with known batch variables should be low and the residual variation high.
- CorNet (a novel systems-oriented algorithm). CorNet is a novel algorithm introduced in this context by the Contact PD/PI John Weinstein and PD/PI Rehan Akbani. It defines a new metric, CZR (pronounced “Caesar”) that quantifies similarities/differences between batches of samples in a network-oriented sense. The algorithm as applied, e.g., to metabolomic data, computes (Pearson or Spearman) correlations between all metabolite-metabolite pairs within batch 1 and the same for batch 2. The result is a vector of correlation values for each batch. Next, it computes the coefficient of determination (r2) between the two vectors. The result is a scalar CZR value that represents the system-wise similarity of the two batches. CorNet is based, not on individual metabolite levels, but rather on the similarities of fully-connected networks generated from the metabolite levels. If two batches are identical, CZR = 1; if one is randomly permuted relative to the other, CZR = 0.
MetaBatch R Package
The MBatch R package is availalbe on GitHub as a direct install to R or as a pre-built DockerHub Container and provided Dockerfile and Docker-Compose files. https://github.com/MD-Anderson-Bioinformatics/BatchEffectsPackage
We will provide MetaBatch as an R-package that programming-savvy users can download to analyze their own data. The R-package will allow users to customize analyses of their own datasets, but its use will require R-language programming expertise well beyond what most biologists and clinical researchers have at their disposal. The data will be processed by the computational back-end of MetaBatch (using R), and results of the analysis will be sent back to the visualization front-end. The user will then be able to download the corrected data and/or save the results of analysis on his/her own machine (or on the cloud). The MetaBatch system will consist of many components that would be complex and tedious to install on end-user machines because of the many libraries and other versioned components required. Therefore, we propose to develop a user-friendly, “ready-to-run” version that non-informatics users can easily install, configure, and use without IT services. It will be “run-anywhere” to avoid the need to support multiple operating systems and user environments. It will be based on Linux container technology using Docker. We were early bioinformatic adopters of Docker and have implemented it successfully for other projects (e.g., NG-CHMs). For users who prefer to download and install MetaBatch on their own machines, we will create an install wizard to enable Macintosh and Windows users to run local instances of the MetaBatch Docker image without expert assistance. Our generic goals:
- A one-stop-shop providing multiple data-quality assessment and visualization algorithms.
- Dynamically explorable plots, with zooming, panning and mouse-over functionalities.
- Novel metrics for interpretation of results.
- Generation of high-resolution graphics for publication.
- No programming experience needed to install and use.
- Flexible adaptable infrastructure for adding algorithms and input/output wrappers for data format conversion.
We will provide technical documentation for bioinformaticians and others whose primary interest is assessing and correcting batch effects. The technical documentation will describe the system architecture and object formats for users who want to develop inter-operable components and/or contribute to the system’s open-source development.
Credits
Research Scientists
- Rehan Akbani
- Bradley M. Broom
- John N. Weinstein
Software Developers
- Chris Wakefield
- Tod D. Casasent
- James M. Melott
- Mary A. Rohrdanz
- Jun Ma
Funding Support
- Weinstein, Broom, Akbani. Computational Tools for Analysis and Visualization of Quality Control Issues in Metabolomic Data, U01CA235510, NIH/NCI